Orbital Tumors
Diagnosis and surgical management of orbital tumors and masses — adult and pediatric, benign and malignant.
Medically reviewed by EyePlastics Medical Editorial BoardASOPRS oculoplastic surgeonsLast updated June 2026
Orbital Tumors
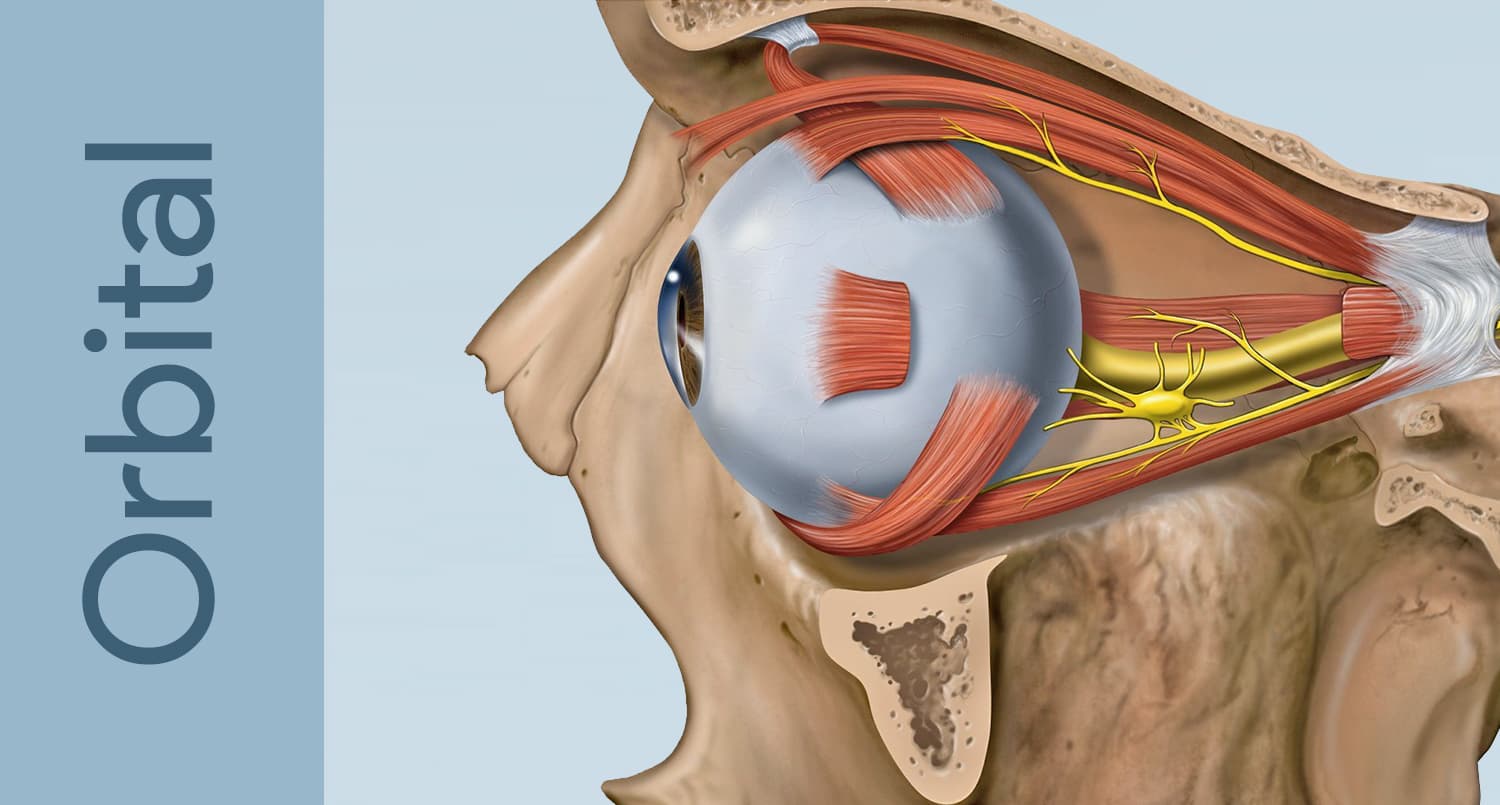
The orbit is a bony cavity approximately 30 mL in volume that houses the eye, six extraocular muscles, the optic nerve, fat, lacrimal gland, and a rich network of blood vessels and nerves. A tumor — any space-occupying lesion, benign or malignant — within this confined space produces characteristic signs by displacing or compressing orbital contents.
Most orbital tumors are benign. The clinical priority is accurate characterization — imaging-guided diagnosis avoids unnecessary surgery for lesions that can be observed, while ensuring timely biopsy and treatment for malignancies. Complex orbital tumors requiring neurosurgical or skull-base approaches are managed in coordination with neurosurgical colleagues.
For a detailed guide to orbital anatomy, see our dedicated Orbital Anatomy page.
Orbital Tumors at a Glance
Anatomy
The orbit is a ~30 mL bony cone holding the eye, six muscles, the optic nerve, fat and the lacrimal gland. A growing tumor has nowhere to expand, so it pushes the eye forward (proptosis) or to the side, and can press on the optic nerve.
How they are evaluated
MRI is best for soft-tissue tumors and the optic nerve; CT shows bone and calcification. Growth rate, location and the patient’s age narrow the diagnosis — many are confirmed with a biopsy.
Adult — most common
Cavernous venous malformation (the most common benign adult orbital tumor), lymphoma, lacrimal-gland tumors, solitary fibrous tumor, schwannoma/neurofibroma and sphenoid-wing meningioma. See adult tumors →
Pediatric — most common
Infantile (capillary) hemangioma, dermoid cyst, lymphatic malformation (lymphangioma) and rhabdomyosarcoma — the most urgent. See pediatric tumors →
Signs and Symptoms of Orbital Tumors
Orbital tumors produce symptoms by displacing the eye or compressing orbital structures. The direction of displacement helps localize the lesion:
- Proptosis (exophthalmos) — forward displacement of the globe; the most common sign of any space-occupying orbital lesion
- Axial vs. non-axial proptosis — lesions within the muscle cone (intraconal) push the eye straight forward (axial); extraconal masses displace the eye away from the tumor
- Double vision (diplopia) — from displacement of the globe or direct infiltration of extraocular muscles
- Visual loss — compression of the optic nerve; may be subtle initially (afferent pupillary defect, color desaturation)
- Pain — more common with rapidly expanding, inflammatory, or malignant lesions; slowly growing benign tumors are often painless
- Eyelid changes — fullness, ptosis, or an obvious mass at the orbital rim
- Palpable mass — particularly for anterior orbital tumors near the orbital rim
Urgent evaluation: Any rapidly progressive proptosis, vision loss, or pain requires prompt CT/MRI imaging and orbital evaluation. Childhood orbital masses deserve urgent assessment — rhabdomyosarcoma grows in days to weeks.
Diagnosis
Diagnosis combines the clinical picture with imaging and, when needed, a biopsy. The patient’s age, the rate of progression, and the presence or absence of pain are powerful clues: a slowly enlarging, painless mass in an adult suggests a benign lesion such as a cavernous venous malformation, whereas a rapidly growing mass — especially in a child — demands urgent evaluation for malignancy.
Imaging is the cornerstone:
- MRI with contrast (and fat suppression) — best for soft-tissue characterization, the optic nerve, and any intracranial or sinus extension.
- CT — best for bone and calcification: bony erosion (malignancy), scalloping or remodeling (benign, e.g. pleomorphic adenoma), hyperostosis (sphenoid-wing meningioma), and fractures.
- Ultrasound helps with anterior lesions and blood flow; CT/MR angiography is used for suspected vascular lesions.
Characteristic patterns often suggest the diagnosis before biopsy — a well-circumscribed intraconal mass (cavernous malformation), soft tissue that “molds” around structures without eroding bone (lymphoma), bony hyperostosis (meningioma), or frank bone destruction (adenoid cystic carcinoma, metastasis).
Biopsy is performed when the diagnosis is uncertain or tissue is needed to guide treatment. Diffuse or infiltrative lesions and suspected lymphoma are sampled by incisional biopsy (lymphoma also needs systemic staging). Well-encapsulated lesions are usually removed whole (excisional). Some lesions should not be biopsied piecemeal — a suspected pleomorphic adenoma of the lacrimal gland is excised intact with its capsule, because rupture risks recurrence and malignant change. Inflammatory presentations get a targeted laboratory workup (including serum IgG4, ANCA, and ACE).
Most Common Orbital Tumors
Age and growth rate narrow the differential before any biopsy. The table shows the most common benign and malignant tumors by age group — follow any entry to its full description on the adult or pediatric page.
| Most common benign | Most common malignant | |
|---|---|---|
| Adults | Cavernous venous malformation (“cavernous hemangioma”) | Orbital lymphoma |
| Children | Dermoid cyst (orbital) · infantile (capillary) hemangioma (periocular) | Rhabdomyosarcoma — an emergency |
Most common in adults
- Cavernous venous malformation — most common overall
- Orbital lymphoma — most common malignancy
- Lacrimal-gland tumors
- Solitary fibrous tumor
- Schwannoma & neurofibroma
- Sphenoid-wing meningioma
Most common in children
- Dermoid cyst — most common overall
- Capillary (infantile) hemangioma
- Lymphatic malformation (lymphangioma)
- Rhabdomyosarcoma — the pediatric emergency
Related Orbital Conditions
Not every orbital mass is a tumor. These related conditions can mimic or accompany orbital tumors:
- Orbital inflammation & pseudotumor (IOIS) — the most common painful orbital mass in adults; treated with anti-inflammatory therapy rather than tumor excision, though a biopsy is sometimes needed to confirm the diagnosis.
- Thyroid eye disease (Graves’ orbitopathy) — the most common cause of proptosis overall; enlarged muscles and fat, not a tumor.
- Fibrous dysplasia of the orbit — a benign expansion of the orbital bone itself.
Warning Signs of an Orbital Tumor
Because the orbit is a closed bony space, a growing mass shows itself by displacing the eye. Signs that warrant imaging include progressive bulging of one eye (proptosis), double vision, a palpable lump, pain, or reduced/dimmed vision from pressure on the optic nerve. New or asymmetric proptosis should always be evaluated.
Imaging & Biopsy
Diagnosis relies on CT and MRI to locate the mass, define its relationship to the optic nerve and muscles, and narrow the differential. Some lesions have a characteristic appearance; others require a biopsy. The most common non-tumor cause of proptosis is thyroid eye disease, which must always be excluded.
Treatment Approaches
Management depends on the diagnosis: observation for stable benign lesions, complete surgical excision for well-defined benign tumors such as cavernous venous malformation (formerly "cavernous hemangioma"), and biopsy followed by tailored therapy (surgery, radiation, chemotherapy, or systemic treatment) for lymphoma and malignancies. Removal is performed through an orbitotomy chosen to reach the tumor with the least disturbance to the eye.
Choosing a Surgeon
Orbital surgery sits at the center of oculoplastic subspecialty practice; an ASOPRS fellowship-trained orbital surgeon works alongside radiology and oncology when needed. Find one in our surgeon directory.
Continue Reading — Complete Orbital Tumor Guide
- Adult Orbital Tumors
- Pediatric Orbital Tumors
- Cavernous Hemangioma
- Capillary Hemangioma
- Orbital Fibrous Dysplasia
Related in-depth guides
- Orbital Lymphangioma (Lymphatic Malformation) — a benign lesion that can suddenly enlarge or bleed.
Frequently Asked Questions
- What are the most common orbital tumors in adults?
- In adults, the most common orbital tumors include cavernous hemangioma (the most common benign primary orbital tumor), lymphoma (the most common malignant orbital tumor in adults), meningioma, dermoid cyst, and lacrimal gland tumors. Metastatic disease from breast, lung, and prostate cancer is also common.
- How are orbital tumors diagnosed?
- Diagnosis begins with a detailed clinical examination and orbital imaging — CT scan (best for bony detail and calcification) and/or MRI (best for soft tissue characterization). For many tumors, biopsy is required for definitive diagnosis. The approach (incisional vs. excisional biopsy) depends on the lesion's location, size, and suspected type.
- What is orbital surgery?
- Orbital surgery encompasses procedures performed within the bony eye socket — including tumor removal, orbital decompression (for thyroid eye disease), orbital fracture repair, and biopsy. Oculoplastic surgeons with orbital subspecialty training perform these procedures, often working with neurosurgery or ENT for complex cases.
- What should I expect during my orbital tumor consultation?
- During your consultation, your oculoplastic surgeon will review your medical history, symptoms, and any imaging studies you've had performed. They will conduct a thorough eye examination, assess your vision and eye movements, and may order additional imaging tests such as MRI or CT scans to better characterize the tumor. Your surgeon will then discuss the findings, explain treatment options tailored to your specific case, and answer any questions you have about the recommended approach.
- What are the main risks and complications of orbital tumor surgery?
- While orbital surgery is generally safe when performed by experienced specialists, potential risks can include changes in vision, double vision, eye movement restrictions, and infection. In rare cases, there may be bleeding, damage to surrounding eye structures, or need for additional procedures. Your surgeon will discuss these specific risks based on your tumor's location and size during your consultation to help you make an informed decision.
- How long is the recovery period after orbital tumor surgery?
- Recovery timelines vary depending on the tumor's location, size, and surgical approach used. Most patients can resume light activities within 1-2 weeks, though complete healing typically takes 4-6 weeks or longer. Your surgeon will provide specific post-operative instructions including activity restrictions, medication use, and follow-up appointment schedules to monitor your healing and visual recovery.
- When should I see an oculoplastic surgeon about a suspected orbital tumor?
- You should seek evaluation from an oculoplastic surgeon if you experience symptoms such as progressive eye bulging, vision changes, eye movement problems, pain, or if imaging has identified an orbital mass. If your general eye doctor or primary care physician has noted concerning findings on examination or imaging, prompt referral to a specialist is important for accurate diagnosis and timely treatment planning. Early consultation is especially critical if malignancy is suspected or if symptoms are rapidly progressing.
- What is the most common cause of a bulging eye?
- The most common cause of one or both eyes bulging (proptosis) is thyroid eye disease, not a tumor. When proptosis is new, asymmetric, or progressive, imaging is done to distinguish thyroid eye disease from an orbital mass.
- Are most orbital tumors cancer?
- No. Many orbital masses are benign -- such as cavernous hemangioma or, in children, capillary hemangioma and dermoid cysts -- and some are inflammatory rather than neoplastic. Treatment ranges from observation to complete surgical removal depending on the specific diagnosis.
Find a Specialist
Connect with a board-certified oculoplastic surgeon who specializes in orbital tumors.
Search the Directory →